
第一作者:Rihui Liang
通讯作者:Wei Feng
通讯单位:北京化工大学先进技术与设备研究所
论文DOI:10.1002/anie.202419165
成果简介
偶氮化合物分子与相变材料通过储存和可控释放光化学和相变能量为可持续能源系统提供了潜在的应用,而开发新型高效的偶氮基太阳能热燃料(STFs)用于光热能量存储以及与有机相变材料的协同合作仍存在重大挑战。本文合成了三种以柔性烷基链为铰链的(邻-、间-、对-)偶氮吡啶聚合物,其中间-偶氮吡啶聚合物表现出惊人的430 J/g的光热存储容量,为开发高能量密度的偶氮基STFs提供了一种可行的解决方案。此外,通过将间偶氮吡啶聚合物与有机相变材料结合,利用氢键和范德华力相互作用共同利用相变能量和光热能量,创新性地构建了稳定的两相混合体系。有机相变材料不仅提供额外的相变潜热,而且还充当溶剂,为偶氮吡啶发色团的光致异构化提供充足的自由体积,这成功地规避了传统偶氮基STFs中冷凝态下的低充电效率和对溶剂辅助充电的依赖。这项研究展示了家庭消费者的能量分配和利用以及光热辅助隔热策略,实现了STFs更广泛的潜在应用。
图文摘要
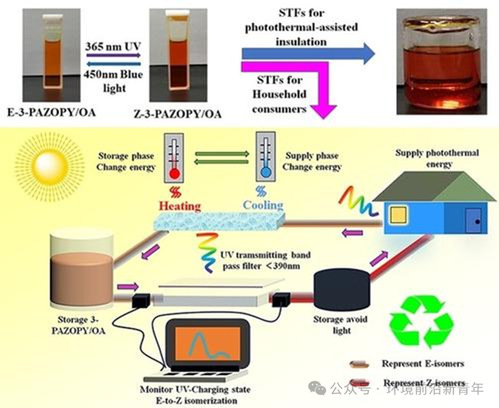
背景介绍
高效利用太阳能已成为减少传统化石燃料消耗和减轻环境污染的重要措施之一,其中最有代表性的方法是光电转换(光伏、光热发电)和光热转换。光热转换通常采用相变材料(PCM)将吸收的光能转换为储存的热能,然而,相变材料受周围环境温度的影响很大,只能储存相变温度以上的热能,这严重限制了太阳热能的利用。另一种利用太阳热能的新方法近年来已经引起了广泛的关注,如已知的太阳热能燃料(STFs)或分子太阳热能(MOST)储能,其利用具有独特的光响应性和光开关的光活性分子,在特定波长的光激发下,伴随着将光热能量储存为化学键,并且储存的光热能量在外部刺激(例如光、热或催化剂)下释放,目前,光致变色化合物包括降冰片二烯-四环烷(NBD-QC),富瓦烯-二钌配合物,二氢薁和偶氮苯等,已广泛应用于短链烷烃中;但NBD-QC合成复杂,产物易降解,这严重限制了在太阳能储存和转化中的实际应用。而偶氮苯由于其简单的结构和可适应的功能化而被广泛研究。鉴于偶氮吡啶分子表现出与偶氮苯发色团相似的可逆光致异构行为,以及作为新型杂环偶氮基STFs的巨大潜力,本文合成了邻位、间位和对位偶氮吡啶聚合物,系统地研究了光致异构行为、光热存储容量和稳定性;最后,通过构建含偶氮吡啶聚合物的绝热器件模型,进一步验证了光热能量在有机相变材料中的有效存储和可控释放。
图文导读
光异构化和热弛豫
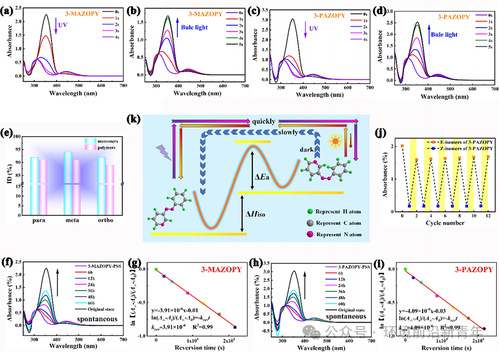
Figure 1. (a) and (c) The time-evolved UV/Vis absorption spectra of 3-MAZOPY and 3-PAZOPY under 365 nm UV light irradiation until reaching thephoto-stationary state. (b) and (d) The time-evolved UV/Vis absorption spectra of 3-MAZOPY and 3-PAZOPY under 450 nm visible light irradiationuntil reaching the photo-stationary state. (e) The E-to-Z photoisomerization degree of azopyridine (para-, meta-, ortho-) monomers and polymers,respectively. (f) and (h) The time-evolved UV/Vis absorption spectra of spontaneous reversion process of 3-MAZOPY and 3-PAZOPY Z-isomers indarkness at room temperature, respectively. (g) and (i) The first-order kinetic Z-to-E isomerization rate constants of 3-MAZOPY and 3-PAZOPY indarkness at room temperature, respectively. (j) Photoisomerization cycling stability of 3-PAZOPY under alternating illumination of UV and visiblelight. (k) The schematic diagram of reversible photoisomerization for azopyridine.
通过紫外/可见吸收光谱监测合成的偶氮吡啶单体和相应聚合物的光致异构化,所有偶氮吡啶单体和聚合物在特定波长激发下表现出一致的变化趋势。对于3-MAZOPY和3-PAZOPY,可以观察到π-π*跃迁吸收带连续降低,同时最大吸收波长蓝移,而n-π*跃迁吸收带在365 nm紫外光照射下连续增加,在5 s内达到光稳态(PSS)(图1a、c);随后,π–π*跃迁吸收带逐渐增加,最大吸收波长回到其初始位置,而n-π*跃迁吸收带在450 nm可见蓝光照射下逐渐减小(图1b、d)。图1e为计算所有偶氮吡啶单体和聚合物的光异构化程度,一般来说,偶氮发色团的亚稳态Z-异构体逐渐克服热障(ΔEa),并由于N=N双键的高扭转应变而自发回复到稳定的E-异构体(图1k)。图1f和1h分别为室温下黑暗中3-MAZOPY和3-PAZOPY随时间变化的UV/Vis吸收光谱。此外,如图1g和1i所示,通过线性拟合确定反向弛豫过程的一级动力学速率常数。可以看出,邻位和间位偶氮吡啶单体和聚合物,比对位偶氮吡啶单体和聚合物显示出更长的半衰期,这归因于苯环对位吡啶中氮原子的强吸电子能力以及烷氧基的给电子能力,导致构成明显的推拉电子结构,从而降低半衰期。而较短的半衰期对于太阳热能储存应用是不利的。从图1j(对于3-PAZOPY)中可以看出,在重复交替的UV和可见光照射循环之后,在所有样品中没有观察到显著的光衰减,这表明所合成的偶氮吡啶单体和聚合物表现出优异的光化学异构化循环稳定性。
光热能量储存

Figure 2. (a) DSC curves of UV-Charged 3-MAZOPY undergoing heat-cooling cyclic at the scanning rate of 5 ℃/min. (b) The schematic ofmechanism for photothermal energy and phase change energy storage in the process of 3-MAZOPY photoliquefaction. DSC curves of UV-Charged4-PAZOPY(c), 3-PAZOPY(d) and 2-PAZOPY(e) undergoing heat-cooling cyclic at the scanning rate of 5 ℃/min, respectively.
图2a为三种偶氮吡啶单体在充入紫外光后的DSC热分析图。研究发现,在第一次加热过程中,与4-MAZOPY和2-MAZOPY相比,只有3-MAZOPY表现出显著的放热峰,ΔHiso为46 kJ/mol;此外,放热峰在第二次加热过程中消失,表明UV-充电的3-MAZOPY通过热诱导模式将所有储存的化学能释放为热量,同时亚稳态Z-异构体完全回复为稳定的E-异构体。值得注意的是,在第一次冷却过程中,在22–60 ℃的范围内观察到一个显著的结晶峰,结晶焓(ΔHc)为50 J/g,这归因于首次加热带紫外线的3-MAZOPY后形成的E-异构体的结晶。总能量密度为180 J/g,3-MAZOPY独特的光液化性质允许同时储存光热能量和相变能量,可以由可见光或加热触发以释放结合的热量,这代表了提高STFs能量密度的有效策略。关于光热能量和相变能量储存以及3-MAZOPY的受控释放的机制的示意图也在图2b中示出,由于半衰期短(τ1/2=2.6 h),在第一次加热过程中没有观察到明显的放热峰。对于2-MAZOPY,放热温度范围与Z-异构体的熔化温度范围重叠,并且在熔化过程中吸收了大量的热流,这掩盖了异构化转变释放的热量。 三种偶氮吡啶聚合物在紫外光照射后的DSC热谱图如图2c~2e所示,所有样品在第一次加热过程中都出现了明显的放热峰,表明光热能量得到了有效的储存。其中,2-PAZOPY的能量密度最低,为30 J/g,其次是4-PAZOPY,能量密度为50 J/g。有趣的是,3-PAZOPY获得了高达430 J/g的能量密度,这对于偶氮基光热存储材料来说是超高水平的。
模型化合物的理论计算

Figure 3. The electrostatic potential (ESP) distribution diagram of iso-surface for E-isomer of the constructed model compounds (the redregion represents positive charge, the bule region represents negativecharge).
计算构建了具有相似给电子和吸电子效应的三种偶氮吡啶单体和更简单的对甲氧基取代的偶氮吡啶化合物(4-MeOAZOPY、3-MeOAZOPY和2-MeOAZOPY)的模型,进行了理论量子化学计算,以阐明热异构化过程中两种异构体的能量变化和光致异构化过程中的分子轨道激发。偶氮吡啶分子模型的E-和Z-异构体的静电势(ESP)分布和偶极矩分别绘制在图3中。可以看出,由于吡啶氮原子的强吸电子效应和烷氧基的给电子效应,4-MAZOPY和4-MeOAZOPY构成了明显的推拉电子结构;而2-MAZOPY和2-MeOAZOPY由于偶氮双键靠近吡啶氮原子而表现出显著的p-π共轭效应,这增强了芳环中π电子的离域。热诱导异构化通常在基态S0实现,过渡态(Ts)通过-C-N=NC二面角旋转定义为异构化机制,这由几何结构优化和频率分析确定。仅发现虚频率,进一步的内禀反应坐标(IRC)分析表明Ts同时连接E-异构体和Z-异构体,证实了Ts和异构化途径的合理性。
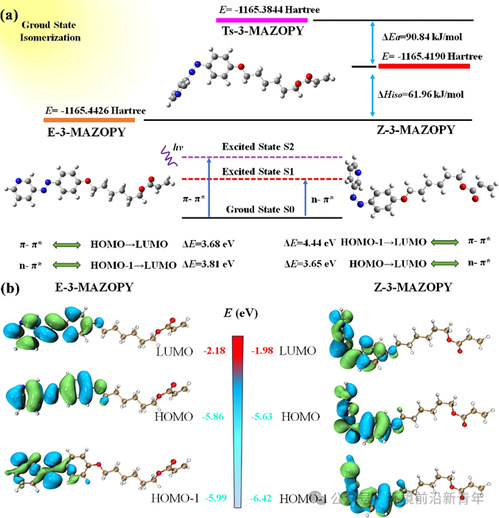
Figure 4. (a) The schematic diagram of electronic energy of E-isomer, Z-isomer and transition state in the ground state S0 as well as the orbitalenergy levels in the excited states for 3-MAZOPY. (b) The molecular orbital (LUMO, HOMO, and HOMO-1) distribution and energy of the 3-MAZOPY E-isomer and Z-isomer by theoretical calculation at function and basis set of standard B3lyp/6-31G (d, p).
更直观、更形象的3-MAZOPY的异构化以及Hiso和ΔEa如图4a所示。可以发现,与4-MAZOPY和2-MAZOPY相比,3-MAZOPY的E-和Z-异构体之间的Hiso和ΔEa最高,而4-MAZOPY的ΔEa最低。这与实验测量的偶氮吡啶单体的ΔEa和τ1/2的变化趋势一致。此外,采用含时密度泛函理论(TD-DFT)分析激发态的分子轨道。在MO框架下,所有偶氮吡啶分子的Z-异构体的LUMO和HOMO能级相对于E-异构体表现出显著的提升,而HOMO-1能级表现出相反的情况(图4b),这是因为Z-异构体中N=N的空间扭曲相互作用更大,导致芳环的π电子离域减少。同时,3-MAZOPY和2-MAZOPY的两种异构体的LUMO、HOMO和HOMO-1能级与4-MAZOPY相比有显著的增加,但是E-和Z-异构体之间的能级之间没有表现出显著的差异。E-异构体的n-π*跃迁(S0→S1)主要涉及从HOMO-1到LUMO的跃迁,而π–π*跃迁(S0→S2)主要涉及从HOMO到LUMO的跃迁;相反,Z-异构体的n-π*跃迁(S0→S1)主要涉及从HOMO到LUMO的跃迁,而π–π*跃迁(S0→S2)主要涉及从HOMO-1到LUMO的跃迁。可以观察到,三种偶氮吡啶单体的E-异构体的π-π*激发能(E-π-π*)几乎相同,而3-MAZOPY的E-π-π*在Z-异构体中位置最低,表明3-MAZOPY的E-和Z-异构体之间的π-π*跃迁吸收带间距最低,ΔE-π-π*为0.72 eV。4-MAZOPY的E-和Z-异构体的n-π*激发能之差(E-n-π*)最大,ΔE-n-π*为0.19 eV,表明n-π*跃迁吸收带实现了有效的分离,这有利于实现可见光双向光开关。
偶氮吡啶聚合物在有机相变材料中的储能和释能

Figure 5. (a) FT-IR spectra of 3-PAZOPY, OA and 3-PAZOPY/OA. (b) and (c) The BS (%) and T (%) intensity plotted as the function of the height of sample tube. The color of the curves represents different time duration of the sample under the isothermal condition of 40 ℃, respectively. (d) The profiles of TSI over time for 3-PAZOPY-10/OA at different temperature of 40 ℃, 50 ℃ and 60 ℃. (e) The time-evolved UV/Vis absorption spectra of 3-PAZOPY/OA under 365 nm UV light irradiation until reaching the photo-stationary state. (f) The time-evolved UV/Vis absorption spectra of 3-PAZOPY/OA under 450 nm visible blue light irradiation until reaching the photo-stationary state. (g) and (h) The DSC curves of OA and 3-PAZOPY/OA for the crystallization and melting temperature measure at a scanning rate of 5 ℃/min, respectively. (i) Degree of supercooling of OA and 3-PAZOPY/OA at different scanning rate of 5 ℃/min, 10 ℃/min, 20 ℃/min and 30 ℃/min.
将高能量密度的3-PAZOPY直接溶解在辛酸(OA)中,创新性地制备了偶氮吡啶聚合物/有机相变复合材料(3-PAZOPY/OA)。可以观察到3-PAZOPY均匀且稳定地分散在OA中,并表现出良好的流动性。通常,由于分子极性的差异,偶氮聚合物很难溶解在OPCMs中,而3-PAZOPY在OA中的溶解度显著提高,因为吡啶氮原子上孤对电子的存在作为受体使得在OA中形成氢键。FT-IR光谱进一步证实了3-PAZOPY很好地溶解在OA中,并且存在氢键,如图5a所示。在3000–3500 cm-1范围内观察到明显的-OH拉伸振动带,表明羧基与羧酸二聚体分离,这可以在OA中930 cm-1处看到明显的吸收峰。这是由于吡啶中的氮原子与羧基之间形成氢键,加速了二聚体的解离和游离羟基的出现。随着OA中3-PAZOPY含量的增加,位于1600 cm-1的芳环伸缩振动带逐渐增强。 通过光学方法估计了在40℃、50℃和60℃的不同温度下3-PAZOPY-10在OA中的稳定性(图5b-c)。研究发现,3-PAZOPY在OA中的反向散射和透射率分别为大约20%和90%,并且随时间和温度增加的变化可忽略,表明3-PAZOPY-10/OA保持优异的透明度和稳定性,没有任何分层。此外,通过Turbiscan软件计算Turbiscan稳定性指数(TSI),得出不同温度下3-PAZOPY在OA中的稳定性,结果如图5d所示,TSI越小,意味着样品的稳定性越好。随着温度的升高,3-PAZOPY/OA的TSI开始降低,然后升高,这可能是由于升高的温度加速了聚合物分子链的运动速率,从而降低了相邻链之间的分子间相互作用,导致聚合物溶胶颗粒的尺寸减小,稳定性增强。偶氮吡啶聚合物在OCPMs中的有效光异构化是作为STFs的重要基础。类似于在UV和可见光照射下3-PAZOPY在溶剂中随时间变化的UV/Vis吸收光谱,3-PAZOPY在OA中的溶液表现出明显的光转换性质,如图5e和5f所示。此外,还测定了室温下黑暗中3-PAZOPY在OA中的热反弛豫过程的一级动力学速率常数,半衰期约为32.3 h。同时,3-PAZOPY/OA在反复交替暴露于UV和可见光后表现出优异的光异构化循环稳定性。 通过DSC以5 ℃/min的扫描速度测定OA的熔融和结晶温度以及相变焓。如图5g所示,当3-PAZOPY被引入到OA中时,结晶温度从9℃下降到约2℃,并且随着3-PAZOPY含量的增加而几乎保持不变。这是因为偶氮吡啶聚合物的引入降低了OA分子排列的有序性和规整性,从而降低了其结晶能力。相反,如图5h所示,随着3-PAZOPY的加入,熔化温度稍微升高(从18℃到19℃),然后稍微降低。此外,所有样品的过冷度(ΔTs=Tf−Tc)和扫描速率之间的关系如图5i所示。较高的过冷度有利于光热能量和相变焓的低温存储。同样,也测定了不同3-PAZOPY含量和扫描速率的带紫外线3-PAZOPY/OA的相变温度和过冷度,结果表明,随着3-PAZOPY含量的增加,UV-荷3-PAZOPY/OA的结晶温度先下降后略有上升,而熔融温度呈持续下降趋势。这是因为紫外照射后,3-PAZOPY的大部分E-异构体转化为Z-异构体,削弱了3-PAZOPY和OA之间的氢键,以及相邻OA之间的分子相互作用,导致熔融温度下降。
光热辅助绝缘
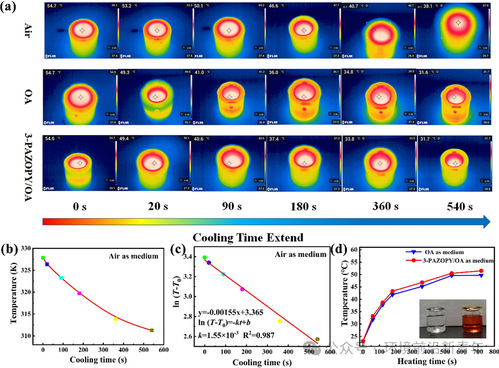
Figure 6. (a) The time-evolved IR thermal image of the water superficial temperature in devices with interlayers of OA, 3-PAZOPY/OA and airduring the nature cooling. (b) The profiles of the temperature of water in the device with interlayers of air vs cooling time. (c) The linear fitting of the cooling rate water in the device with interlayers of air by Equation S5. (d) The profiles of the surface temperature of water in devices with interlayers of OA and 3-PAZOPY/OA vs heating time. The inserted is the optical photograph of the devices with interlayers containing OA (left)and containing 3-PAZOPY/OA (right).
在制备的3-PAZOPY/OA的辅助下,搭建简单隔热装置可延迟热量损失,该装置由真空绝缘层和STFs层构成,STFs层在通过远程控制光照射以帮助真空绝缘层延迟热损失来释放光热能量中起重要作用。将具有相同质量和初始温度的去离子水置于具有OA、3-PAZOPY/OA和空气(空气代替真空)夹层的装置中。如图6a所示,在自然冷却过程中,使用红外热成像相机定期监控去离子水的表面温度。通过拟合冷却曲线(图6b),发现作为绝缘介质的空气在整个冷却温度范围内完全符合线性关系,如图6c所示;而OA和3-PAZOPY/OA在不同的冷却温度范围内也表现出良好的线性拟合;为了进一步比较它们的冷却速率,计算OA和3-PAZOPY/OA在整个冷却温度范围内的平均冷却速率,可以得出两者的冷却速率相对接近,而空气的冷却速率最低(在实际的真空环境中,由于缺少传热介质,冷却速率甚至会更低)。 由于实验研究中使用的可见光通常来自LED点光源,无法在大面积上提供全面的照明,因此采用了热诱导方法。此外,在上述具有OA和3-PAZOPY/OA夹层的装置中,水的冷却速率接近,这意味着传热性能非常相似。因此,采用OA和3-PAZOPY/OA作为传热介质对加热过程中水的温度变化的影响可以忽略不计。夹层含有3-PAZOPY/OA的装置通过365 nm紫外照射,随后将具有相同初始温度和质量的水放入其中,并观察相同加热过程中的温度变化。图6d为加热过程中装置中水的表面温度变化的红外热像和相应的温度-时间关系曲线,可以观察到,在相同的加热时间下,含有3-PAZOPY/OA的装置获得了比OA更高的水温;此外,接近加热期结束时,OA装置中的水温保持相对稳定,而3-PAZOPY/OA装置中的温度由于热刺激诱导的光热能量释放而持续升高。
结果展望
总之,本文设计并合成了三种类型的(邻位、间位和对位)偶氮吡啶单体及其相应的柔性烷基链聚合物,其中,间偶氮吡啶单体表现出显著的光解行为,为无溶剂STFs的发展奠定了重要基础;此外,间偶氮吡啶聚合物表现出稳健的光热储能稳定性(τ1/2约为47.1 h)和优良的储能密度(Hiso约为430 J/g),为开发新型高能量密度偶氮基STFs开辟了新的途径。同时,利用含活泼氢原子的有机相变材料与偶氮吡啶聚合物之间的氢键和范德华力相互作用,直接构建了均匀稳定的偶氮基有机相变复合材料两相混合物体系。与常规偶氮苯有机相变复合材料相比,其优势在于,有机相变材料不仅提供潜热储存,而且作为偶氮吡啶聚合物双向光异构化的溶剂,能够有效储存和可控释放光热能量,而不需要外部溶剂辅助。此外,通过讨论偶氮吡啶聚合物掺杂量和扫描速率对熔融和结晶行为的影响,实现了对光热和相变能量存储和释放的精确控制。在这种考虑下,本文提出了一个家庭消费者的能量分配和利用系统的概念和光热辅助绝缘策略,其可行性通过关于光热的存储和控制释放行为的证明来验证。这项研究也为智能窗、除冰、建筑和绝缘涂层等潜在应用提供了一个重要的设计框架。

